Page History
...
Usługa UNRES jest przyjazną dla użytkownika realizacją pakietu modelowania gruboziarnistego białek, opartego na polu siłowym UNRES opracowanym w wyniku współpracy grupy prof. Adama Liwo z Wydziału Chemii Uniwersytetu Gdańskiego oraz grupy prof. Harolda A. Scheragi, Cornell University, USA. Dzięki znacznemu uproszczeniu reprezentacji łańcucha polipeptydowego przy jednoczesnym ścisłym związku wyprowadzonego pola siłowego z fizyką oddziaływać symulacje są przyspieszone 1000 - 10000 razy w stosunku do obliczeń w reprezentacji pełnoatomowej. Adresatami usługi są biofizycy, biologowie molekularni i bioinformatycy, którzy mają potrzebę symulowania zmian strukturalnych, dynamiki i termodynamiki dużych układów białkowych w czasie rzeczywistym. W szczególności usługa będzie przydatna dla badaczy, którzy chcą wykonać wspomniane wielkoskalowe symulacje białek natomiast brak czasu nie pozwala im na zapoznanie się z uruchamianiem zadań bezpośrednio na serwerach przy użyciu systemu kolejkowania.
...
- Obliczanie i lokalna minimalizacja energii cząsteczki białka.
- Kanoniczne symulacje gruboziarniste dynamiki białek.
- Symulacje białek przy użyciu metody wymiany replik lub zwielokrotnionej wymiany replik i konstrukcja na tej podstawie profili zależności wielkości geometrycznych, mechanicznych i termodynamicznych od temperatury, badanie krajobrazów energii swobodnej białek, termodynamiki zwijania, itp.
- Przewidywanie struktury białek oparte o fizykę oddziaływań.
Aktywowanie usługi
Co należy aktywować, aby móc skorzystać z usługi? (Założenie konta, certyfikat, grant?, aktywacja konkretnych usług w portalu). Należy pamiętać o istnieniu rozdziałów ogólnych podręcznika, do których warto się odwołaćNależy złożyć wniosek o dołączenie do zespołu plggunresng (zespół użytkowników usługi UNRES).
Ograniczenia w korzystaniu (podsekcja opcjonalna)
...
Koniecznie z przykładowymi zrzutami ekranu lub fragmentami kodu.
Zaawansowane użycie
Przewidywanie de novo struktury białka przy pomocy pola siłowego UNRES
Pole siłowe UNRES umożliwia przeprowadzanie przewidywania de novo struktury białka, korzystając jedynie z sekwencji aminokwasowej. W tym celu należy wykonać symulację dynamiki molekularnej ze zwielokrotnioną wymianą replik (MREMD) wprowadzając sekwencję białka o poszukiwanej strukturze do przykładowego (Rysunek 1) pliku wejściowego MREMD.
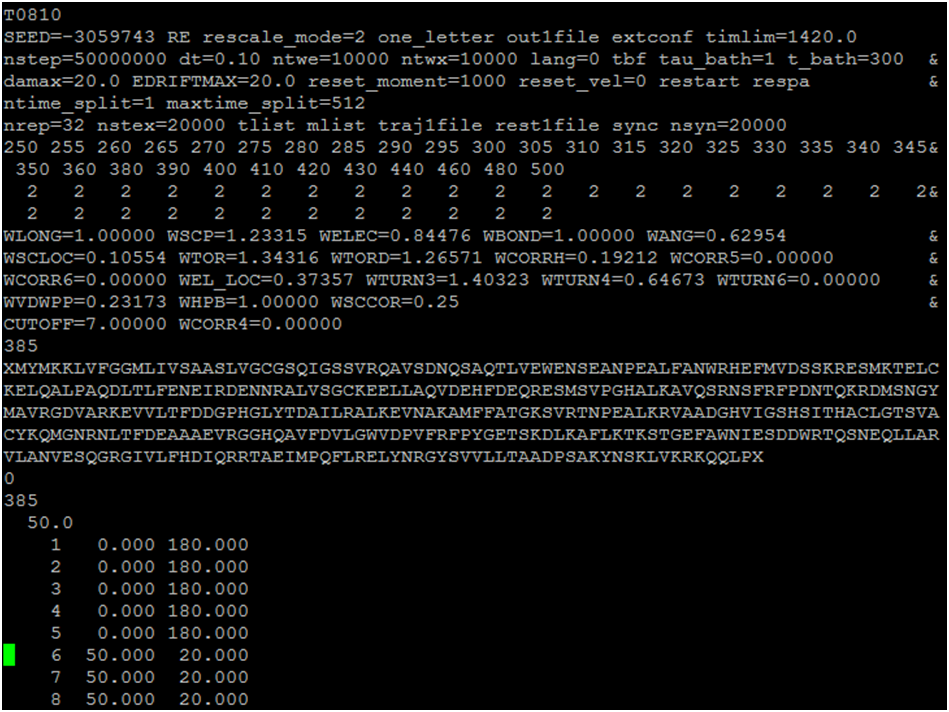
Rysunek 1. Przykładowy plik wejściowy używamy w polu siłowym UNRES. Linia pierwsza stanowi komentarz, linie 2-14 zawierają parametry używane w polu siłowym, linia 15 zawiera całkowitą liczbę reszt aminokwasowych symulowanego układu z uwzględnieniem „dummy atom”, natomiast kolejne linie zawierają sekwencję aminokwasową w kodzie jednoliterowym wraz z „dummy atom” (litery X). Ostatnie linie wykorzystywane są do wprowadzenia ograniczeń swobody układu (więzów) np. na strukturę drugorzędową lub odległości między parami atomów.
Przy wprowadzaniu sekwencji należy pamiętać o dodawaniu „dummy atom” na początku i końcu każdego łańcucha białka (litery X w kodzie jednoliterowym).
W celu przyspieszenia symulacji można wprowadzić do pliku wejściowego dane o strukturze drugorzędowej (przewidzianej przy pomocy zewnętrznego narzędzia, takiego jak PSIPRED czy JPred) w postaci listy wartości brzegowych kątów torsyjnych ograniczających swobodę łańcucha (ostatnie linie pliku wejściowego).
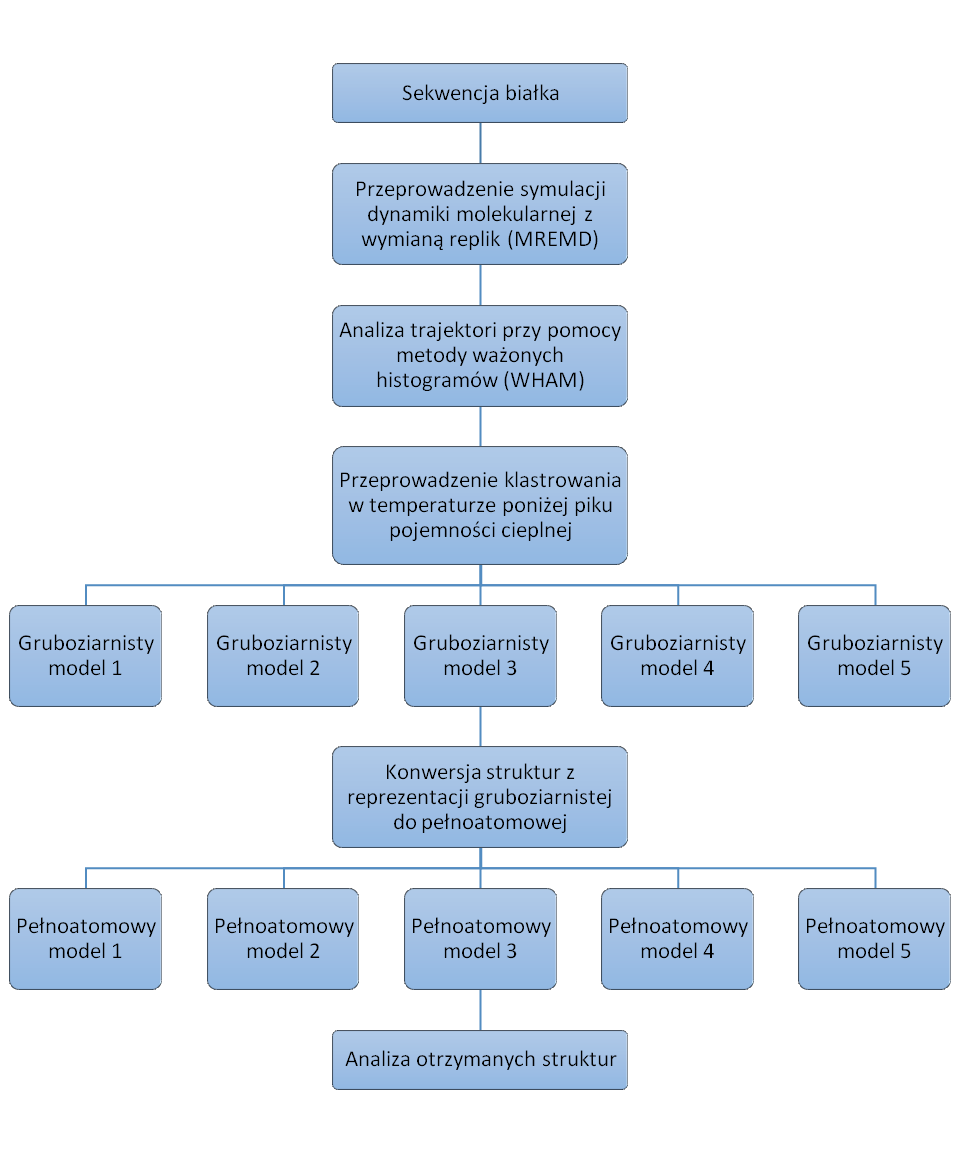
W celu przeprowadzenia analizy otrzymanych trajektorii należy użyć podprogramu WHAM, wykorzystującego metodę analizy ważonych histogramów do określenia prawdopodobieństwa wystąpienia struktur w danych temperaturach. Następnie konieczne jest przeprowadzenie analizy skupień (klastrowania) przy użyciu podprogramu Cluster, w temperaturze poniżej temperatury piku pojemności cieplnej, w celu wytypowania najbardziej prawdopodobnych struktur białka. Otrzymane w ten sposób struktury należy następnie przekonwertować z postaci gruboziarnistej do pełnoatomowej przy użyciu dostępnych narzędzi (np. programu Pulchra).
Zalecany schemat postępowania został umieszczony na poniższym diagramie:
 Ewentualnie jako osobny podrozdział.
Ewentualnie jako osobny podrozdział.
Gdzie szukać dalszych informacji?
Strony zewnętrzne (jeśli są), odnośnik do helpdesku lub strony dokumentacji o pomocyStrona dokumentacji pakietu UNRES ze szczegółowym opisem wprowadzanych danych (w języku angielskim).
| Info |
|---|
| Można też dodać sekcję "Co dalej?" ze wskazaniem (odnośnikiem) do dalszej części dokumentacji, o ile jest wymagana. |
...
Overview
Content Tools